

|Articles|March 8, 2018
Pearls and pitfalls for principal investigators
Author(s)Mark Packer, MD, FACS, CPI
Interested in becoming a principal researcher? Read this first.
Advertisement
Editor’s Note: Welcome to “
In his latest blog, Mark Packer, MD, FACS, CPI, shares the rewards of becoming a principal investigator (PI) in clinical research, as well as the common mistakes to avoid. There are many reasons an ophthalmologist might want to be involved in clinical research as a PI:
- Being on the cutting edge of innovation is exciting.
- Clinical research raises the profile of the practice and the PI.
- Involvement in scientific advancements means contributing to the improvement of patient care and outcomes.
- If managed correctly, clinical research can be a lucrative adjunct to clinical practice.
- Your patients will receive the latest innovations long before they are available to the general public (while, of course, taking some additional risks).
Over the course of many years’ involvement in clinical research as a PI, medical monitor and subject matter expert, I have observed some PIs who outperform their peers and others who create problems for their sponsors. I’d like to share a few of the practices that make a PI successful.
1) Be scrupulously honest on the site selection questionnaire
As a surgeon, one learns to “underpromise and overdeliver" with patients to avoid unrealistic expectations and the inevitable subsequent dissatisfaction. The same holds true for PIs and their communications with sponsors. I don’t recommend intentionally underestimating your site’s enrollment capability, but I do recommend avoiding overestimating. In the face of uncertainty, err conservatively. Almost nothing will stretch a sponsor’s patience thin like failing to meet enrollment goals.
If you are new to clinical research, it may be difficult to provide an accurate enrollment estimate. However, one can analyze recent patient records to determine how many would meet the inclusion/exclusion (I/E) criteria.
For example, in an IOL study, the sponsor is usually looking for subjects with healthy eyes other than cataract (i.e., no zonulopathy, no glaucoma, no macular degeneration, no retinopathy). It’s easy enough to look through last quarter’s cataract patients and find those without comorbidity.
Of course, not every eligible patient will want to participate in a clinical trial. I would subtract all those who opted for presbyopia-correcting or toric IOLs, and then take 25% of the remainder as a conservative estimate.
2) Do not try to make yourself look good at the technology’s expense
Some complications cannot be avoided. Every clinical trial has adverse events (AEs). We should not try to minimize their importance, but it is not a good idea to brag about them either. For example, I have seen PIs present their management of surgical complications related to investigational devices at conferences without first consulting the sponsor.
Irrespective of whether that constitutes a breach of confidentiality (it does), I’m not sure what purpose it serves other than to increase the notoriety of the PI at the sponsor’s expense.
If one wants to participate in future research projects, I would treat everything that occurs within the context of a clinical trial as confidential. In fact, the protocol will often contain a paragraph that constrains investigators from presenting or publishing data without the sponsor’s consent.
3). A case report form (CRF) is not a case report article
In medical school, we learn to record all the positive findings and all the pertinent negative findings. The CRF is a list of only those history and examination fields determined relevant to the clinical trial, and only those findings that fit in each of the fields need to be recorded.
However, there is almost always an area for additional comments. Some PIs want to add a lot of detail, much as they would for a case report in a medical journal. Not only is this unnecessary, it can lead to queries and questions from regulatory authorities that complicate the review process without adding any significant information.
For example, some subjects may have a lot to say about their visual perception following cataract or refractive surgery, and they may use a vocabulary all their own. These subjective impressions are best captured through a validated questionnaire, so that their significance can be evaluated consistently among subjects and across trials. It’s not helpful to write down every adjective a patient may use to describe a halo.
4). Know your expenses
The clinical trial agreement (CTA) between the PI and the sponsor will spell out the compensation for conduct of the study, usually in terms of subjects and form completion. To effectively negotiate a fair agreement, the PI (or designee) must know what it will cost the site to run the trial per subject. An experienced site manager will know the cost for every test and procedure required by the protocol, as well as the costs associated with approval from an institutional review board (IRB), administration, surgery center services, and the PIs’ time.
If a sponsor’s proposal doesn’t net a profit, one may still decide to participate in a study if the value of participation includes important intangibles-at least one will know why one is participating.
On the other hand, one may wish to participate in a lucrative, but not particularly exciting trial to enhance the site’s bottom line. In either case, these decisions cannot be made in the absence of knowledge.
5). Understand the regulatory environment
It’s impossible to do a good job as a PI without a thorough understanding of the regulatory process and its requirements. Every site initiation visit (SIV) includes a review of good clinical practice (GCP) and the protection of human subjects. However, the PI’s understanding should go beyond these basics.
What are the relevant regulatory time lines related to starting the study, obtaining review and achieving approval? What is involved in a site inspection? What have been the challenges encountered by the sponsor during previous investigations of similar devices or drugs? The greater your understanding, the better job you can do in helping the sponsor reach the goal.
This list of pearls and pitfalls is not exhaustive by any means. But it is a start, whether you are just getting involved in clinical research or seeking to improve your performance as a PI.
Newsletter
Don’t miss out—get Ophthalmology Times updates on the latest clinical advancements and expert interviews, straight to your inbox.
SubscribeAdvertisement
Related Content
Advertisement


Latest CME


















Advertisement
Advertisement
Trending on Eye Care Network - Ophthalmology Times
1
FDA grants RMAT designation to Emmecell's EO2002 for corneal endothelial disease
2
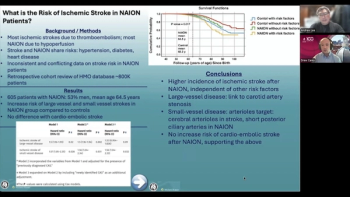
NeuroOp Guru: What a big data study reveals about NAION and stroke risk
3
Port Delivery System Therapies in Retinal Vascular Diseases
4
Opus Genetics reaches FDA alignment on phase 3 registrational design for OPGx-LCA5 gene therapy
5