

Commentary|Videos|November 22, 2023
NeuroOp Guru: Novel mitochondrial mutation found in a case of isolated LHON
Author(s)Andrew G. Lee, MD, Andrew Carey, MD
Andrew Lee, MD, and Andrew Carey, MD, sit down on another episode of the NeuroOp Guru to discuss a novel mitochondrial mutation found in a case of isolated LHON (Leber hereditary optic neuropathy).
Advertisement




















Video Transcript
Editor's note - This transcript has been edited for clarity.
Andy Lee, MD:
Hello and welcome to another edition of the NeuroOp Guru. I'm here with my good friend Drew Carey. Hi, Dr. Carey.
Drew Carey, MD:
Hi Andy.
Andy Lee, MD:
And today we're gonna be talking about a novel mitochondrial mutation found in a case of isolated LHON, Leber hereditary optic neuropathy. And so Drew, maybe you could just give us a little background on why this is important and what is the meaning of it?
Drew Carey, MD:
So, Leber hereditary optic neuropathy is a genetic condition of mitochondrial inheritance. So it's passed down through the maternal line. And not everybody who carries the gene will have vision loss. It depends on the gene and some other environmental features, and possibly some gender predilection for who manages to manifest vision loss and who doesn't. We classically, think of the three primary Leber mutations that account, at least in folks of Western European heritage, accounts for probably 95% ofthe cases. But as we're discovering that more patients who present with the Leber hereditary-like pattern of optic neuropathy with sudden vision loss, either simultaneously or rapid sequentially in both eyes. It's painless and central scotomas, and they don't recover with steroids and you don't have any enhancement on MRI scan. There are other genes that appear to behave very similarly to what we consider the 3 primary genes. And particularly if you're not of Western European descent, the chances that one of those classic primary genes as the cause drops off precipitously. And so as we're trying to do a better job of A - diagnosing patients and as treatments become available, treating them. Also giving them prognostic information for the patient and their family members, it's important that we are able to genotype patients and understand, you know, their genetic molecular basis for their condition. As well, it's particularly difficult sometimes to know if a mitochondrial variant is pathogenic or not. There's a lot of variability in mitochondrial genes, and not all changes are pathogenic. And in a condition like Leber where we have incomplete penetrance. It's difficult to say, well, you have this gene and you have vision loss, but your mom has it, and she doesn't have vision loss. That's totally consistent with Leber, but in an entirely different type of condition. You would say, well, that means that can't be the gene that causes the vision loss. So it's important we collect these cases and get a better understanding, so we can be better diagnosticians. And eventually, treaters for this condition.
Andy Lee, MD:
So I find this pedigree to be quite fascinating. It's like a female, and then she has these 3 daughters, and they're unaffected. And then all of a sudden, there's these 2 affected males that appear in generation 3. Can you explain to our audience how that could happen? Why would you have 3 unaffected females and then female to male transmission? And how is that compatible with mitochondrial disease?
Drew Carey, MD:
Absolutely. So the mitochondria are inherited through the maternal line. When sperm fertilizes an egg, something happens to the mitochondria in the sperm, and they don't get passed along to the new person. So it's only the maternal mitochondria that get passed along. In in the vast majority of of LHON, it's this mitochondrial gene that's really important for the electron transport chain for respiratory metabolism. That is affected and most the time it's one of the NADH dehydrogenase subunits. And not everybody who carries the gene is impacted. Men, classically, it's said about 50% of those who carry the gene will have vision loss, and in women only 10%. And there's some environmental factors. Smoking increases your risk of conversion, cocaine use, being extremely dehydrated. Other metabolic, toxins or metabolic stressors may increase your risk of a vision loss as they stress this mitochondria. And so when you look at a pedigree like this, if you weren't thinking mitochondrial. You might say, Oh, that looks like maybe it's autosomal dominant with incomplete penetrance. The real clue for mitochondrial conditions, is there's no male to male transmission. It always has to be down the maternal line. So men have a predominance, but it has to be passed along through a maternal line. And so if you didn't have the the, the maternal grandmother here, you might think, oh, it could be recessive or X-linked. But this pedigrees, is very classic for mitochondrial disease. And sometimes the big kicker is if there's a maternal uncle. Now there's no, there's no males in the second line there that are descended from the grandmother. But if you had a maternal uncle, who's involved that that's a real clue in the pedigree as well.
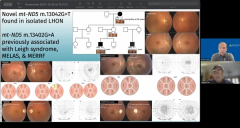
Andy Lee, MD:
Mitochondrial mutation here was 13402G>A, and that's like, MELAS and MERRF and Leigh syndrome and how could that be? You have different phenotypes, Mitochondrial encephalomyopathy, lactic acidosis (MELAS) and Myoclonic epilepsy with ragged red fibers (MERRF). These are completely different phenotypes from LHON, and yet they have the same genotype. That's so strange.
Drew Carey, MD:
Yeah and that's one of the things that we are just beginning to really understand about mitochondrial disease. You know, there's a lot of diseases that we - LHON is a classic example of what's called a polygenic disease, where you can have more than one gene that develops the same phenotype. And we see that with our three primary mutations, in LHON. They all look the same. And you wouldn't be able to say, this is the gene for this patient with LHON, without doing the genotyping. And they could look exactly the same as another person. In mitochondrial disease and some nuclear diseases as well, we're starting to realize that a single gene can have multiple phenotypes, and that's called pleiotropy. And, particularly in mitochondrial disease, it's perhaps a little bit more common, where it's not a one-to-one ratio of genotype to phenotype. And a lot of it has to do with the fact that these genes are present in every tissue. And they are really responsible for energy production. Right, the mitochondria is called the "powerhouse" because they do that electron transport chain that's responsible for developing most of the ATP and high energy tissues like nerves, and cardiac tissue, and skeletal muscle. And there's probably modifying factors in with other genes that may have to have influence on the severity of disease in which tissues are involved. Leigh syndrome is our most severe mitochondrial disease, it's usually onset in infants. They have a multi-organ failure. Ophthalmic manifestations can include nystagmus, ophthalmo-plegia, in addition to optic atrophy. As well as systemic manifestations, including neurologic things like hypotonia. Metabolic manifestations like lactic acidosis. Cardiac manifestations, hypertrophic cardiomyopathy, and they have extremely short lifespans, although there's milder versions of Leigh that may have later onset. And then MELAS and MERRF are kind of more mild, multiorgan, manifestations of mitochondrial disease. And we generally think of LHON as isolated, although we have elite LHON+, which may have little pieces of all these different mitochondrial manifestations. And we don't have a complete understanding of why patients may present with one phenotype versus the other and there may be other mitochondrial genes or nuclear genes that interact with these genotypes to really give us a phenotype. But I think that's why it's important to keep a broad differential of your genotype [inaudible] condition when you see a patient. Just because they don't have one of the 3 primary LHON genes doesn't mean it's not a mitochondrial disease or does not mean it's LHON or LHON-like. So if they're not from the classic western heritage, Western European heritage, you may want to start off with full mitochondrial sequencing rather than just the 3 primary mutations. And you may have better chances of getting genotyping in your your first testing round.
Andy Lee, MD:
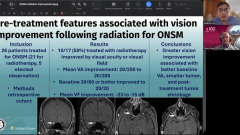
And it looks like all these those OCTs are pretty much the redzone and all the fields are showing Central scotomas bilaterally. Is that kind of the take home from the clinical standpoint, if we see a bilateral simultaneous or sequential central scotoma, with optic nerve that looks like this. Might have Pseudopapilledema in the beginning but then become optic atrophy that we should be ordering the mitochondrial testing and the LHON mutations on these types of patients.
Drew Carey, MD:
Yeah, I definitely think you want to keep it in the differential, you know, if the history does not really tell you that they've been exposed to a typical toxin that might do this. Or they don't have ethambutol or disulfiram. They don't have, you know, a clear dietary deficiency. You know, they're not vegan, we probably still want to check them for B-12, because it's a treatable and reversible condition, potentially reversible, and it's going to have other neurologic deficits if you don't catch it and treat it. But I definitely think if you know, if you have unexplained optic atrophy, where it's simultaneous or rapidly sequential with central scotomas, you definitely want to think about mitochondrial disease [inaubdible]. Dominant optic atrophy is going to be slowly progressive from childhood, a lot of patients just kind of think that's the way their vision always is. These patients have normal vision for very long portion of their life and then have a sudden change. And that's a big tip off that that's mitochondrial rather than dominant optic atrophy, or even one of our recessive optic atrophies.
Andy Lee, MD:
What do you think the take home message should be Drew for our audience in terms of this novel, mitochondrial mutation in LHON?
Drew Carey, MD:
Yeah, so I think you know, the big take home points are unexplained optic atrophy - think about genetics. If you have a family history that appears to run on the maternal line, think about mitochondrial. Don't stop at your 3 primary mutations if you think it's LHON. Or if patients aren't coming from a typical Western European heritage, maybe start with mitochondrial full sequencing, rather than just your 3 primary mutations. And in a lot of labs it's the same price or maybe even cheaper just to do mitochondrial sequencing, deletion duplication testing at the start.
Andy Lee, MD:
That's fantastic Drew. Thanks again. And that concludes another edition of the NeuroOp Guru.
Drew Carey, MD:
Thanks, Andy.
Advertisement
Related Content
Advertisement



Latest CME


















Advertisement
Advertisement